
NEWS RELEASE 12-JUN-2024
Peer-Reviewed Publication
TSINGHUA UNIVERSITY PRESS
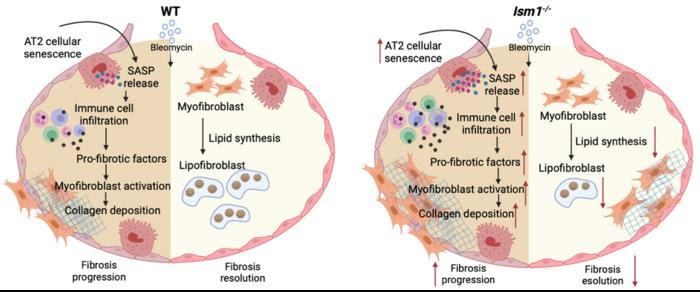
IMAGE:
DURING FIBROSIS PROGRESSION, ISM1-/- MICE EXHIBITED ENHANCED CELLULAR SENESCENCE IN THE ALVEOLAR TYPE II EPITHELIAL (AT2) CELLS WITH INCREASED RELEASE OF SENESCENCE ASSOCIATED PRO-INFLAMMATORY FACTORS WHICH LEAD TO ENHANCED IMMUNE CELL INFILTRATION, RELEASE OF PRO-FIBROTIC MEDIATORS PROMOTING THE ACTIVATION OF MYOFIBROBLASTS WITH EXCESSIVE COLLAGEN DEPOSITION, RESULTING IN ENHANCED BLEOMYCIN INDUCED PULMONARY FIBROSIS (BIPF). DURING FIBROSIS RESOLUTION, REDUCED LIPID SYNTHESIS GENES AND DECREASED TRANS-DIFFERENTIATION OF MYOFIBROBLASTS TO LIPOFIBROBLASTS LEADS TO DELAYED FIBROSIS RESOLUTION IN ISM1-/- MICE. THIS FIGURE WAS GENERATED USING BIORENDER.
CREDIT: RUOWEN GE, NATIONAL UNIVERSITY OF SINGAPORE
Idiopathic pulmonary fibrosis (IPF) is a chronic and progressive lung disease marked by the thickening and scarring of lung tissue with unclear etiology. Affecting around five million people worldwide, IPF causes severe respiratory problems and greatly diminishes the quality of life. Despite ongoing medical research, the exact cause of IPF is still unknown, and treatment options are limited. The prognosis for IPF is grim, with only about 20% of patients surviving five years post-diagnosis, highlighting the critical need for better therapies and a deeper understanding of the disease.
The histopathological features of IPF include honeycomb cysts, fibroblastic foci, proliferative epithelial cells and accumulation of excessive extracellular matrix (ECM). However, the detailed cellular and molecular dynamics within the lungs during IPF progression remain under active investigation and new effective therapeutic options need to be explored.
In this article, the authors used bleomycin-induced pulmonary fibrosis (BIPF) mouse model, which is commonly employed in IPF research to understand the pathophysiological mechanisms of the disease and to explore new therapeutic options. The BIPF mouse model undergoes three phases: 1. The inflammatory phase. Immediately following the initial damage of lung epithelial cells by bleomycin, immune cells rush into the lung and release pro-inflammatory factors necessary for fibroblast activation. 2. The fibrotic phase. The fibroblasts in the lungs become activated and convert to myofibroblasts that produce excessive collagen, causing scarring in the lung. 3. The resolution phase. Myofibroblasts transform into another type of cell called lipofibroblasts which help to reduce the fibrosis and heal the lung.
In this study, the researchers identified that a lung abundant secreted protein called ISM1 acts as an endogenous protective factor against pulmonary fibrosis using BIPF model and Ism1 knockout mice (Ism1-/- mice), a genetically engineered mouse which is deficient in ISM1.
First, the authors show that under normal conditions, ISM1 deficiency altered lung gene expression signature to project an increased susceptibility to pulmonary fibrosis. Next, they showed that ISM1 deficiency exacerbates lung fibrosis in BIPF. During the fibrotic phase, Ism1-/- lung exhibited increased cellular senescence (cell aging) of alveolar type II epithelial cells (AT2) and fibroblasts, leading to enhanced release of pro-inflammatory factors. This led to increased immune cells infiltration into the lung, thereby triggered the enhanced expression of factors necessary for conversion of fibroblasts to myofibroblasts, which further resulted in excessive collagen deposition in the Ism1-/- mice. During the fibrosis resolution phase, absence of ISM1 led to reduced lipid synthesis gene expression accompanied by decreased conversion of myofibroblasts to lipid-laden lipofibroblasts. Lipofibroblasts are known to facilitate the resolution of pulmonary fibrosis.
Overall, this study further indicates that ISM1 is an anti-inflammatory protein in lung and provides new insights into its protective role in resisting pulmonary fibrosis. It underscores the importance of ISM1 during both the progression and resolution phases of pulmonary fibrosis. Thus, further research on how to manipulate ISM1 level in pulmonary fibrosis is warranted for the exploration of new intervention/treatment options.
This work is supported by grants awarded to Ruowen Ge from Singapore Ministry of Education (partially supported by grants A-8001134-00-00 and MOE-T2EP30221-0011). The funding agency was not involved in the study design, data collection, analysis, interpretation, writing of this manuscript, and the decision to submit the article for publication.
See the article:
Ism1 deficiency in mice exacerbates bleomycin-induced pulmonary fibrosis with enhanced cellular senescence and delayed fibrosis resolution https://doi.org/10.1016/j.hlife.2024.05.006
JOURNAL
hLife
DOI
10.1016/j.hlife.2024.05.006
ARTICLE TITLE
Ism1 deficiency in mice exacerbates bleomycin-induced pulmonary fibrosis with enhanced cellular senescence and delayed fibrosis resolution
ARTICLE PUBLICATION DATE
31-May-2024
Leave a Reply