
DOI:https://doi.org/10.1016/j.ajhg.2023.09.011
Summary
Advances in long-read sequencing and assembly now mean that individual labs can generate phased genomes that are more accurate and more contiguous than the original human reference genome. With declining costs and increasing democratization of technology, we suggest that complete genome assemblies, where both parental haplotypes are phased telomere to telomere, will become standard in human genetics. Soon, even in clinical settings where rigorous sample-handling standards must be met, affected individuals could have reference-grade genomes fully sequenced and assembled in just a few hours given advances in technology, computational processing, and annotation. Complete genetic variant discovery will transform how we map, catalog, and associate variation with human disease and fundamentally change our understanding of the genetic diversity of all humans.
Introduction
We are quickly moving past the celebration of the first release of a complete, telomere-to-telomere (T2T) assembly of a human genome to now anticipating the production release of hundreds, if not thousands, of completely sequenced human genomes. The development of completely sequenced human T2T chromosomes represented a seed change in the field of human genetics because, in principle, all forms of variation could be discovered irrespective of class, frequency, or location. The state of the art up to that point involved the production of reference genomes organized into contigs where gaps corresponded to some of the most highly repetitive and structurally complex regions of the genome. Key to resolving these was the development of long-read sequencing. Long-read sequencing and assembly, while still led by a small number of specialized sequencing centers,1
has made it possible to construct phased assemblies from diploid genomes. Though currently more expensive than short-read sequencing, the costs continue to drop rapidly and throughput is increasing. With the anticipated gains in genome technology, reaching complete T2T-sequenced human genomes is expected to become more accessible and routine. What will this mean for how we map, catalog, and associate variation with human disease? How will access to complete genome sequencing alter the foundation of the future of precision medicine? And how can we broaden access to the benefits of this emerging technology?
We propose that over the next ten years, technological gains in long-read sequencing technologies will deliver routine access to T2T-phased genome assemblies. Ready access and democratization of these complete assemblies from researchers and clinicians around the world will be impactful in expanding variant discovery, understanding genetic diversity, and broadening epigenetic characterization. The potential for T2T genomes to completely capture the full spectrum of human genetic variation is beginning to transform the field of human genetics in terms of both our comprehension of genetic variants and their functional impact and how we represent and associate this variation with human disease.
Access to complete genomes will improve genetic association with human health and disease
At its most fundamental level, human genetics focuses on establishing the link between genotype and phenotype. The discovery of genetic variation, in turn, has always been dependent on advances in technology. Although cytogenetics, microarrays, optical maps, and short-read sequencing (e.g., Illumina and other sequencing platforms) all provided access to different classes and types of genetic variation, no method has been comprehensive in terms of the types, classes, or sizes of variants it can discover. Illumina whole-genome sequence provides, for example, cost-effective and accurate access to single-nucleotide variants (SNVs)4
for 85%–90% of the genome. Larger genetic variation classes, including specific repeat regions like segmental duplications (and the genes therein), larger mobile elements, centromeres, sites of heterochromatic secondary constriction (i.e., chromosomes 1qh, 9qh, 16qh, and Yqh), and acrocentric portions of human chromosomes, have been largely overlooked in variation studies. This is due to their improper assembly in the human genome and the challenges of unambiguously hybridizing arrays or mapping short-read sequences (<250 bp), which are shorter than the repeats themselves. This has translated into a limited understanding of human genetic variation and diversity and underlies some of the missing heritability of disease for Mendelian and complex genetic traits.5 Long-read sequencing technologies, such as nanopore sequencing from Oxford Nanopore Technologies (ONT)6 and single-molecule, real-time (SMRT) sequencing technology from Pacific Biosciences (PacBio),7 are transforming the field of human genetics. First, they increase the sensitivity for the detection of structural variant changes (insertions, deletions, and inversions >50 bp) by 2- to 3-fold.8
Structural variants are 3-fold more likely to underlie a genome-wide association study (GWAS) signal and 50-fold more likely to affect the expression of a gene than an SNV.10
This means that variants of larger effect are being disproportionately uncovered as genomes are sequenced with long reads. Second, both ONT and PacBio involve the sequencing of native DNA (as opposed to amplified material) where CpG methylation can be readily distinguished. This so-called “fifth base” allows epigenetic differences between individuals and tissue types to be readily identified, leading to the discovery of new disease mechanisms that associate genetic variants with DNA modifications. Finally, both the length and accuracy of current long-read sequencing have meant that phased genome assemblies can now be generated where both the maternal and paternal haplotypes are resolved nearly telomere to telomere.14
Furthermore, this can now be done in the absence of parental data by assessing methylation status available from long-read sequence data from imprinted loci within the phased genome assembly.15
This has had the transformational effect that we no longer think of the human genome as 3 Gbp but rather 6 Gbp where all variants are fully sequence resolved and phased with respect to all other genetic differences on that haplotype.
Genetic imputation is, in principle, no longer required to reconstruct haplotypes and previously inaccessible regions of our genome (Figure 1) can now be resolved and accessed for genetic and epigenetic variation. This includes the sequence resolution of complex regions, such as SMN1 and SMN2, important targets for gene therapy associated with spinal muscular atrophy (SMA)16
; the complete resolution of CGG repeat expansions in the fragile X syndrome gene FMR1, as well as the repeat’s methylation status; and the precise length and composition of the Kringle IV repeat domain of lipoprotein A, the major genetic risk factor associated with coronary heart disease and stroke risk particularly in individuals of African American descent.17
In addition to these biomedically relevant regions, long thought to be off limits to sequence, assembly, and genetic association, we are now resolving the sequence and variation of other more complex segmental duplications, acrocentric chromosomes,19 and centromeres22
for the first time. The study of the short arms of the human acrocentric chromosomes offers insight into shared, large homologous regions, including ribosomal DNA repeats and near-identical blocks of segmental duplications and satellite DNA.19
High-resolution maps of these regions can reveal pseudo-homologous regions between short arms,19
identifying arrangements that are compatible with crossover in inverted duplications near previously reported breakpoints of Robertsonian translocations,23
and support new models of ongoing recombination exchange.21
Additionally, T2T assemblies have opened the door to base-level studies within centromeric regions,22
which has allowed the scoring of satellite DNA variants26
and the precise epigenetic mapping of CpG base modifications27
and centromere-protein enrichment,22
resulting in detailed models of array evolution at sites of kinetochore assembly.22
Access to high-resolution maps of satellite arrays will advance evolutionary studies of rates of non-homologous crossover and conversion and also require the development of methods29
to confidently align satellite arrays and perform meaningful variant calling. In doing so, low-resolution labels such as “heteromorphisms,” traditionally defined cytogenetically by visible gains and losses of satellite DNA staining, will become enhanced as we adopt a more meaningful definition of the underlying genomic structure necessary to understand centromere identity and function and its role in chromosomal aneuploidies. Overall, the ability to read genomes completely will offer new insight into missing heritability and new models of mutation and establish a new standard of comprehensive genetic association with human health and disease.
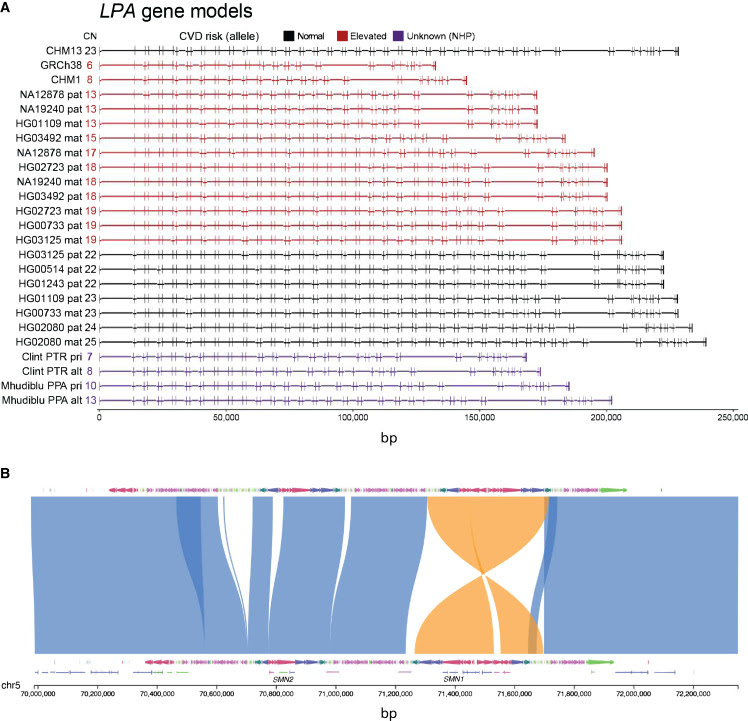
Figure thumbnail
Figure 1Sequence resolution of complex, biomedically relevant human genetic variation
Routine access to complete genomes will require another step change in technologies
The cost, throughput, and computational infrastructure to routinely create and analyze T2T-phased genomes are still currently rate limiting. The mechanics of how to do so, however, have largely been solved over the last few years—a combination of more accurate long-read sequencing with extremely long read lengths (>100 kbp) has led to the development of new assembly algorithms that can now phase and assemble most of the human genome if sufficient high-molecular-weight DNA can be generated. Verkko14
and hifiasm1
both couple the accuracy of high-fidelity (HiFi) PacBio data31
to create the backbone of large sequence contigs with the scaffolding potential of ultra-long ONT data32
to assemble the maternal and paternal complement of chromosome arms and, in some cases, entire chromosomes.33
Moreover, both long-read sequencing platforms continue to improve throughput, increase accuracy, and reduce costs. The release of the PacBio Revio sequencing platform has been estimated to increase throughput by more than 5-fold, while ONT continues to develop new nanopores and duplex sequencing to improve sequencing accuracy.34
At present, we estimate the underlying sequencing costs for generating 30-fold sequence coverage from each of the two long-read sequencing platforms to be less than $5,000. If including labor, computational infrastructure, as well as additional ancillary technologies, such as Hi-C needed for phasing, one can generate nearly complete phased genomes with fewer than 100 gaps for ∼$10,000–15,000. It remains to be seen whether best practices for T2T genomes will continue to involve the use of both platforms or if a single platform will emerge. It is clear, however, that costs will drop and throughput is increasing, thus making phased genomes a possibility for basic research pursuits.
Beyond the mechanics, a greater challenge facing human geneticists is to accurately represent the complexity of human genetic variation being uncovered. Unlike the bulk of human genetic variation (i.e., SNVs), which involve the change of a single base from one nucleotide to another, much of the newfound variation being uncovered is more complex in nature involving structural changes, copy number changes of entire genes, interlocus gene conversion, or even the complete evolutionary turnover of large swathes of portions of the genome (e.g., centromeres, acrocentric short arms).21
While many tools have been developed in the last couple of years to visualize36
and understand the evolutionary dynamics of such regions, cataloging these differences between human haplotypes and recording them in the form of a standard VCF is a particular challenge. Placing such variation into the context of human haplotypes, however, is a huge advantage that will be made possible by the emergence of more sophisticated genotyping tools to facilitate genotyping of these and other complex regions of the genome.37
As studies shift from one to hundreds of genomes,39
it is clear that one reference genome is an incompletely sufficient baseline for comparison and understanding the complexity of human variation.
A pangenome reference resource will represent common and shared haplotypes
There is a need to modernize the human reference genome to better reflect the full range of genomic diversity across the globe. In doing so, this new reference resource will contain a more accurate and diverse representation of global genomic variation, improve disease-association studies across populations, expand the scope of genomics research to the most repetitive and polymorphic regions of the genome, and serve as the ultimate genetic resource for future biomedical research and precision medicine. Genetic variations between individuals can impact the effectiveness of medical treatments and influence the risk of developing certain diseases. By creating a more diverse genome reference, we can better understand these variations and develop more personalized and effective medical treatments. Additionally, the diversity of human genomes reflects the complex history of human migration and evolution. By including a more diverse range of genomes in the reference, we can gain a more comprehensive understanding of human history and evolution. To address this challenge, the Human Pangenome Reference Consortium aims to develop and release a new human reference resource that is defined by hundreds of aligned common human haplotypes (i.e., observed with an allele frequency of 1%), known as “pangenome.” This new pangenome reference will be an important foundation for identifying and predicting the functional outcomes of variants across diverse populations.
There are two stages to the development of a new human pangenome reference. The first involves the production of a series of nearly complete haplotype-resolved genomes where all genes and genetic variants are represented linearly and the diversity of human genetic variation is adequately surveyed. The second involves the development of methods to represent this diversity where the shared parts are distinguished from those that are variable (Figure 2) and, as a result, all subsequent human genomes can be mapped better than they would to a single reference genome.41
The goals are simple: eliminate the reference bias introduced by mapping to a single reference and, thereby, improve the mapping and genotyping accuracy of genomes more distantly related to that single reference. A particularly popular approach in the field of computational genomics has been attempting to develop graphical models of the pangenome.42
While details on construction and visualization vary, multiple sequence alignments essentially undergird pangenome graphs where the variation between any two linear haplotypes is represented as “bubbles” of various sizes and the individual linear haplotype sequences can be reconstructed as a path or a walk through the graph (Figure 2). The first human pangenome graph using 47 human genomes (94 haplotypes) was recently constructed using three different approaches allowing variation in more structurally complex loci (e.g., HLA, RHD, CYP2D6, etc.) to be better represented and characterized.39
Importantly, the results showed that the pangenomic approach outperformed all other approaches not only for the characterization of structural variation but also for smaller variants, including single-nucleotide polymorphism and indels, significantly decreasing errors and improving accuracy genome-wide. The routine generation of human genomes is thus an essential endeavor, as additional genome data augment the pangenome’s robustness and precision, ensuring it remains the most effective tool for mapping, genotyping, and interpreting human genetic variation.
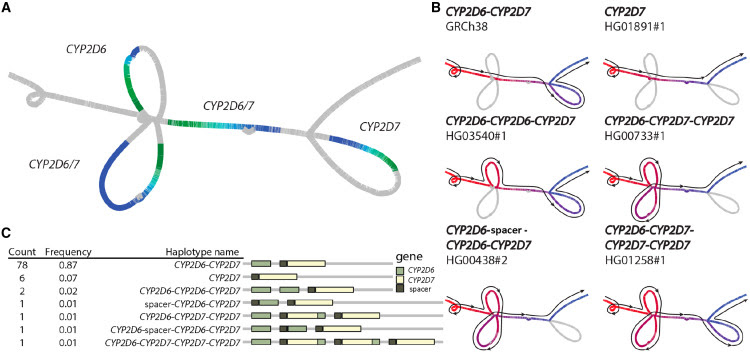
Figure thumbnail
Figure 2A graph-based representation of structural variation of the CYP2D6/D7 locus
Bold predictions
In this perspective, we have taken the position that added value of complete genetic information will make T2T genomes of individuals an inevitability for both basic and clinical research. Given this, we conclude by making five bold predictions with respect to its impact on our field.
Understanding new mutational mechanisms and their relationship to human health
Routine and comprehensive human genome sequencing and mapping are poised to revolutionize our understanding of mutational mechanisms and their impact on human health. Three such mechanisms are ectopic exchange for acrocentric DNA,21
recombination processes that drive satellite evolution in centromeric DNA,28
and interlocus gene conversion of segmental duplications.30
Acrocentric DNA, which contains repetitive sequences at the ends of chromosomes, is prone to ectopic exchange—a process where non-allelic homologous sequences recombine. This can result in chromosomal rearrangements and has implications for genetic disorders, such as Robertsonian translocations. Similarly, segmental duplications, which are sizable, identical DNA sequences found in different locations within the genome, can undergo interlocus gene conversion, a non-reciprocal transfer of genetic information between the duplicated segments. This process can alter gene function or regulation and change susceptibility to genomic disorders by homogenizing large swathes of sequence to be 100% identical in specific human haplotypes. More recently, additional mutational mechanisms associated with segmental duplications have become apparent from the generation of phased genome assemblies, namely, an increased SNV mutation with a transversion bias of cytosine to guanosine in segmental duplications when compared to unique DNA30
and recurrent inversion toggling as a result of non-allelic homologous recombination between inverted segmental duplications.
In contrast, satellite DNA found enriched in human centromeres evolve extensively through mechanisms of intrachromosomal exchange that result in haplotype-specific variation.44
The interchromosomal exchange between repeat copies of shared homologs is less frequently observed, and interchromosomal exchange between non-homologs is expected to be rare.22
Changes in the satellite array structure spanning known regions of kinetochore assembly and centromere function can impact chromosome segregation during cell division.46
Therefore, advancing studies of the mutational mechanisms of centromeric satellite arrays offer new insight into studies of aneuploidy,47
a condition associated with various disorders such as Down syndrome, Edwards syndrome, and certain cancers.
As we move toward regular, complete genome mapping, our capacity to detect, study, and understand these event
Leave a Reply