
by Johns Hopkins University School of Medicine
Unexpected heterogeneity of ribosomal RNA genes in human populations revealed by genome studies suggests potential variation in protein translation by the ribosomes. Credit: Wenjun Fan, Ph.D.
A genome study undertaken by Johns Hopkins Kimmel Cancer Center researchers to look for variants in a gene considered a fundamental building block for microscopic structures that synthesize proteins took a surprising twist.
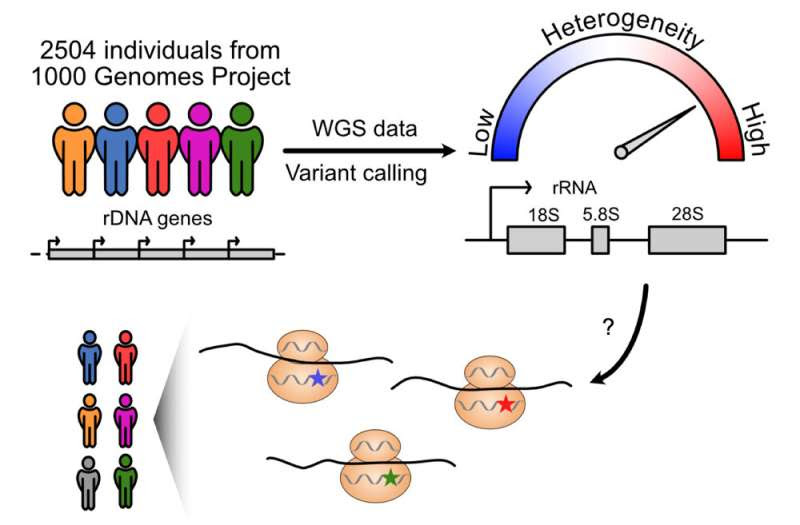
Human ribosomal RNA (rRNA) genes are essential for building ribosomes, or machineries that translate proteins. The study findings, to be published in the Feb. 2 issue of the journal RNA, demonstrated that these genes—which were thought to be similar among people—instead differed significantly based upon a person’s geographic ancestry. Particularly high variations were found on a segment called 28S rRNA, an essential component of the protein-translating ribosome.
The team, directed by Marikki Laiho, M.D., Ph.D., director of molecular radiation sciences in the Department of Radiation Oncology and Molecular Radiation Sciences, veered from their regular research focus of developing new molecules that could be potentially useful in the treatment of cancer to delve into a basic biology concept they wanted to better understand.
They had developed cancer drugs that target synthesis of ribosomal rRNAs, a unique process that drives cancer development. Without these, cancer cells cannot multiply. The team wondered if the rRNA gene itself was altered in cancers, and how that could affect their targeting strategy. Despite the importance of this gene, there was no definitive reference sequence published to date.
Team members set out to take a bioinformatics approach to rRNA gene sequences, using high-performance computers at the Maryland Advanced Research Computing Center, a joint venture managed by The Johns Hopkins University and the University of Maryland. To start charting cancer alterations, they had to understand whether variants existed in the human population. The rRNA gene sequence was considered “untouchable,” or so fundamental that it seemed unlikely to have any variations.
“However, when we started that analysis, we very quickly realized that the cancer genomes were highly aberrant,” Laiho says. “In order for us to understand whether that aberration is real—meaning that it changes in a particular cancer—we needed to better understand what a normal human gene looks like.”
Next, they used whole-genome sequencing data from the 1000 Genomes Project (an international human genetics database) to analyze variants in 2,504 individuals from 26 populations. They identified 3,791 variant positions on the rRNA gene. This included 470 variant positions seen on 28S rRNA. Most of these variants were located on long protruding folds of the rRNA that vary among species. These represent positions of diversity, and are potentially under continuous evolution.
“The analysis results were beyond our imagination,” Laiho says. “We saw perfect conservation of sequences across vast swaths of the gene, and then highly variable sites in the exact locations that we expected to be unaltered. This suggests that the way variant rRNAs are built into the ribosomes could lead to potential alterations in how the ribosomes function.”
Many of the variants observed were segregated by population. For example, some variants were far more frequent among African or Asian populations versus American or European populations, and vice versa. This raises the possibility that some of the variants are ancient, ancestry-dependent, yet have been retained in modern populations, Laiho says.
“It’s too early to speculate what these variants mean, but what is fascinating is that they are conserved by population, and this means their retention is somehow important,” she says.
The study findings suggest there is a need to functionally assess how the 28S rRNA variants affect ribosome functions, which could in turn help lead to more targeted therapies for cancer or other diseases, Laiho says.
Leave a Reply