
by Perelman School of Medicine at the University of Pennsylvania
Credit: Cell (2023). DOI: 10.1016/j.cell.2023.11.019
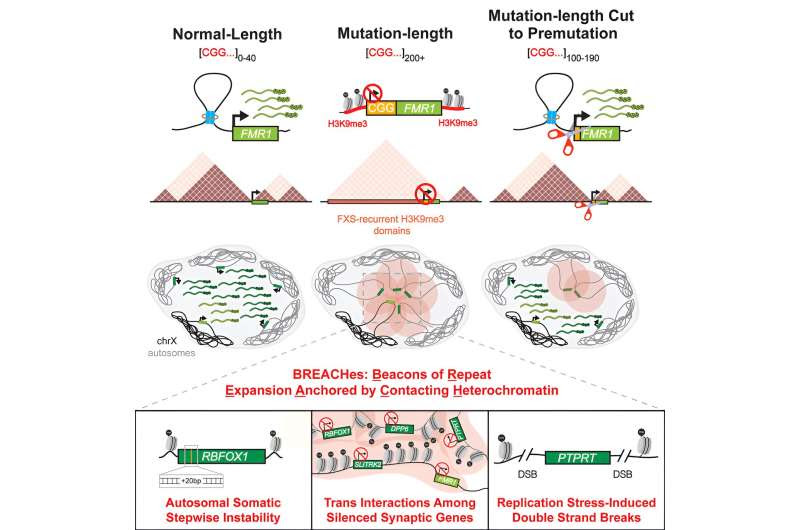
Researchers have found new disrupted genes and an unexpected molecular pattern—dubbed BREACHes—related to fragile X syndrome (FXS), a genetic disorder estimated by the Centers for Disease Control and Prevention to impact about 1 in 7,000 males about 1 in 11,000 females.
The study, by researchers at the Perelman School of Medicine at the University of Pennsylvania, which used cells and brain tissue donated by patients, also showed that simply editing the length of the abnormal repetitive pattern could restore the silenced genes on multiple chromosomes. The study was published in the journal Cell.
“Our findings have implications for future fragile X syndrome treatment strategies and highlight potential mechanisms contributing to genome instability that may underlie other diseases as well,” said study co-first author Linda Zhou, MD, Ph.D., a clinical resident of Dermatology at Penn Medicine.
A team led by senior author Jennifer Phillips-Cremins, Ph.D., an associate professor in Bioengineering and Genetics, and a member of the Epigenetics Institute at Penn Medicine, investigated FXS, the most common form of inherited intellectual disability, in order to add understanding of the disorder’s underlying cause. Textbook models attribute it to the silencing of a single gene, FMR1, and the loss of the protein FMR1 encodes, Fragile X Messenger Ribonucleoprotein (FMRP).
It is widely thought that the loss of FMRP causes severe dysregulation of synapses, which connect neurons in the brain, as well as the disruption of how genes are expressed in neurons’ nuclei. The leading model of FXS was built on studies using a transgenic mouse in which the FMR1 gene was knocked-out. However, the mouse model was missing the critical genetic driver of FXS: a mutation called a “repeat expansion,” which occurs when a long repetition of a sequence of two or more DNA letters grows unstable and abnormally long (a mutation-length repeat).
For FXS, this is the three-letter sequence—CGG—appearing at one end of the FMR1 gene. While a normal version of FMR1 has 40 or fewer CGG triplets in the repeat tract, an FXS patient will have 200 triplets or more. The abnormality triggers a defensive response by the cell, which essentially silences FMR1 and FMRP. Because the repetitive sequence is difficult to engineer, small animal models of FXS lack the repeat tract, and therefore may not have demonstrated important aspects of the role of repetitive DNA in mechanisms underlying FXS.
In their new study, the research team used an array of advanced sequencing and imaging techniques, as well as human cell lines and brain tissue with the CGG repeat expansion, to uncover surprising new patterns of genome disruption in FXS. The researchers discovered that large swaths of multiple chromosomes in FXS patient samples—which include the CGG repeat—are marked with the silencing heterochromatin. These heterochromatin “domains” are coined BREACHes—Beacons of Repeat Expansion Anchoring Contacting Heterochromatin.
BREACHes come together into physically contacting clusters in the nucleus and silence genes involved in neuron synaptic connections, along with genes tied to the integrity of connective tissue such as skin and joints. Disruption to synapses and connective tissue are observed in FXS patients in the clinic, therefore the ability to identify BREACHes has the possibility of being a powerful tool for finding potentially important disrupted genes beyond FMR1.
The researchers also tested whether the repeat could be directly linked to BREACHes by using CRISPR-Cas gene-editing technology to cut the CGG expansion down to a non-FXS-causing length.
“When we cut CGG to a shorter length called premutation (100–190 triplets), we observed that many of the large swaths of silencing heterochromatin were reversed, and multiple chromosomes spatially disconnected from FMR1,” said co-lead-authors Ken Chandradoss, Ph.D., and Ravi Boya, Ph.D., both post-doctoral researchers in Phillips-Cremins’ lab.
The team’s experiments demonstrated that genes originally silenced by BREACHes were re-expressed in FXS cells with the CRISPR-shortened CGG repeat.
“The broad impact of our finding that the mutation-length CGG expansion is necessary for the maintenance of BREACHes is that repeat engineering alone can potentially be used as a therapeutic approach to reverse genome-wide silencing of multiple critical genes potentially contributing to FXS clinical presentations,” said co-lead author Thomas Malachowski, a Ph.D. student in Cremins’ lab.
Future FXS treatments might explore the replacement of the functions of some of the silenced genes identified in the study, not just FMR1. The researchers noted, however, that a more ambitious strategy would be to cut back the excessively long CGG repeat expansion at a defined time in development to prevent or at least reverse the effects of silencing heterochromatin.
Exploring this possibility would need to carefully balance the positive effects of re-activating important genes with the protective role heterochromatin has on guarding against instability of the repetitive genome.
Examples of other disorders potentially impacted by these findings include Huntington’s disease and amyotrophic lateral sclerosis (Lou Gehrig’s disease), which are members of the same, broader class of repeat expansion disorders as FXS, which have been thought to be driven by mutation of a single repetitive tract in the DNA.
Phillips-Cremins also explained that the team observed BREACHes in other human cellular models of genome instability, including with cell lines containing mutations found in cancer or lab-induced DNA breakage.
“Our results suggest that BREACHes may be found in the future to have broader impact on gene silencing in other diseases with genome instability, including certain cancers and other repeat expansion disorders,” she said.
More information: Thomas Malachowski et al, Spatially coordinated heterochromatinization of long synaptic genes in fragile X syndrome, Cell (2023). DOI: 10.1016/j.cell.2023.11.019
Journal information: Cell
Provided by Perelman School of Medicine at the University of Pennsylvania
Leave a Reply