
by University of California, Los Angeles
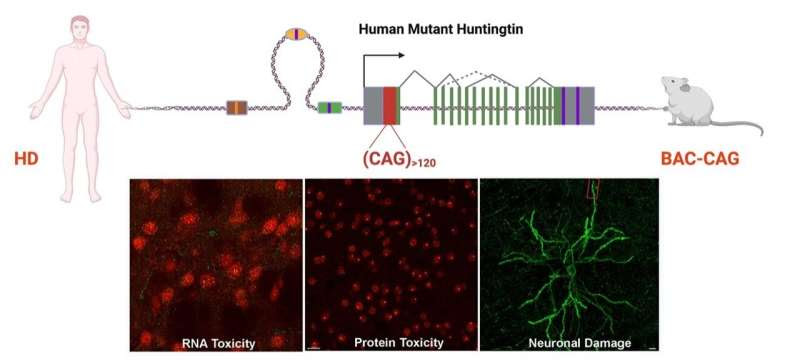
The upper portion of the schematic illustrates generation of a novel mouse model of Huntington’s disease with long, uninterrupted CAG repeats. In the new model, the striatum shows RNA and protein toxicities originated from the pure CAG repeats, which lead to neuronal damage. Credit: UCLA Health/Yang Lab
Researchers at the Jane and Terry Semel Institute for Neuroscience and Human Behavior at UCLA and the David Geffen School of Medicine at UCLA have developed a new mouse model of Huntington’s disease that recapitulates more Huntington’s disease-like characteristics than earlier models. It is providing new clues to the mystery surrounding how genetic mutations dictate disease onset and giving researchers a powerful new tool to test new therapies engaging multiple targets to treat the devastating neurological disorder.
Huntington’s disease affects more than 30,000 people in the U.S., according to the National Institute of Neurological Disorders and Stroke, causing a variety of symptoms, such as personality changes, impaired judgment, unsteady gait and involuntary movements, and speech and swallowing impairment. Although it usually begins between ages 30 and 50, an earlier onset (under age 20) or later onset (after age 70) can occur.
Huntington’s disease is a familial neurodegenerative disorder in which a child of parents with the disease has a 50-50 chance of inheriting the causative mutated gene, named huntingtin. In people who do not have Huntington’s disease, the gene typically contains about 18 repeats of DNA letters C-A-G, but those with this disease may have 40 repeats or many more; the longest stretches found in patients so far contain more than 100 CAG repeats.
“Since Huntington’s disease is caused by a single gene mutation, conceivably it should be easier for therapeutic intervention. However, even though this mutation was found about 30 years ago and scientists around the world fight very hard to find disease-modifying treatment, so far, all the efforts are yet to be successful, especially with the halting of last year’s promising clinical trial to lower mutant huntingtin expression that was a setback to the HD community,” said Dr. X. William Yang, professor of psychiatry and biobehavioral sciences and the Terry Semel Chair in Alzheimer’s Disease Research and Treatment at the David Geffen School of Medicine at UCLA.
This study by the Yang Lab was designed to answer a genetic mystery in Huntington’s disease. Previous studies in the field had focused on the toxic protein products encoded by the CAG repeats, a string of amino acid residues (glutamine) that are toxic to neurons. However, recent human genetic studies with thousands of HD patients revealed an unexpected finding: Patients with CAA interruptions (CAA also encodes glutamine) in the CAG repeats have a later onset of the disease compared to patients without such interruptions but with the same glutamine repeat.
“In this study, we developed the first human genomic transgenic mouse model of Huntington’s disease with long—about 120—uninterrupted CAG repeats and compared the new model to our previous HD model with frequent CAA interruptions. Together, they showed that the long CAG repeat is selectively toxic to the striatum, the brain region that controls movement and cognition and is the most affected in Huntington’s disease,” Yang said.
Yang, senior author of an article published online on Feb. 2, 2022, in the journal Neuron, added that among other findings, the study provides evidence that the new model with long CAG repeats may be toxic at the DNA, RNA and protein levels in brain regions affected by Huntington’s around the time of disease onset.
The new mouse model has a subset of Huntington’s disease-like behavioral deficits, such as motor deficits and sleep disorders, and other characteristics that are largely absent in previous mouse models carrying the human huntingtin gene, such as pathological changes in non-neuronal cells and broad dysregulation of gene expression in the HD-vulnerable brain region.
“Our new model is unique from a therapeutic perspective as it has the entire human huntingtin gene, including several DNA variants present in the patients, and it has a long and pure CAG repeat,” said first author Dr. Xiaofeng Gu, a project scientist in the Center for Neurobehavioral Genetics at the Semel Institute who was primarily responsible for engineering and characterizing the mouse model.
The new model developed in the Semel Institute at UCLA can be used to test candidate therapies to lower the human huntingtin and those targeting the toxicities originated from the pure CAG repeats in huntingtin, said Yang, adding that it also can be used to test combinatory therapies against both types of targets. Currently, the new model has already been used by two pharmaceutical companies and several academic labs to test their therapeutic interventions.
Leave a Reply