
F. Perry Wilson, MD, MSCE
In 1986, in Britain, cattle started dying.
The condition, quickly nicknamed “mad cow disease,” was clearly infectious, but the particular pathogen was difficult to identify. By 1993, 120,000 cattle in Britain were identified as being infected. As yet, no human cases had occurred and the UK government insisted that cattle were a dead-end host for the pathogen. By the mid-1990s, however, multiple human cases, attributable to ingestion of meat and organs from infected cattle, were discovered. In humans, variant Creutzfeldt-Jakob disease (CJD) was a media sensation — a nearly uniformly fatal, untreatable condition with a rapid onset of dementia, mobility issues characterized by jerky movements, and autopsy reports finding that the brain itself had turned into a spongy mess.
The United States banned UK beef imports in 1996 and only lifted the ban in 2020.
The disease was made all the more mysterious because the pathogen involved was not a bacterium, parasite, or virus, but a protein — or a proteinaceous infectious particle, shortened to “prion.”
Prions are misfolded proteins that aggregate in cells — in this case, in nerve cells. But what makes prions different from other misfolded proteins is that the misfolded protein catalyzes the conversion of its non-misfolded counterpart into the misfolded configuration. It creates a chain reaction, leading to rapid accumulation of misfolded proteins and cell death.
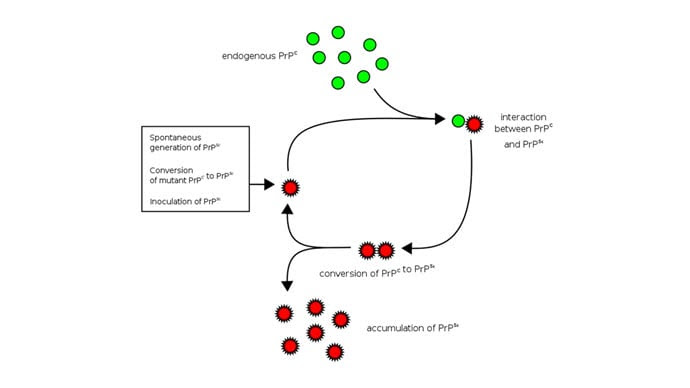
And, like a time bomb, we all have prion protein inside us. In its normally folded state, the function of prion protein remains unclear — knockout mice do okay without it — but it is also highly conserved across mammalian species, so it probably does something worthwhile, perhaps protecting nerve fibers.
Far more common than humans contracting mad cow disease is the condition known as sporadic CJD, responsible for 85% of all cases of prion-induced brain disease. The cause of sporadic CJD is unknown.
But one thing is known: Cases are increasing.
I don’t want you to freak out; we are not in the midst of a CJD epidemic. But it’s been a while since I’ve seen people discussing the condition — which remains as horrible as it was in the 1990s — and a new research letter appearing in JAMA Neurology brought it back to the top of my mind.
Researchers, led by Matthew Crane at Hopkins, used the CDC’s WONDER cause-of-death database, which pulls diagnoses from death certificates. Normally, I’m not a fan of using death certificates for cause-of-death analyses, but in this case I’ll give it a pass. Assuming that the diagnosis of CJD is made, it would be really unlikely for it not to appear on a death certificate.
The main findings are seen here. Since 1990, there has been a steady uptick in the number of deaths due to CJD in this country, as well as an increase in overall incidence.

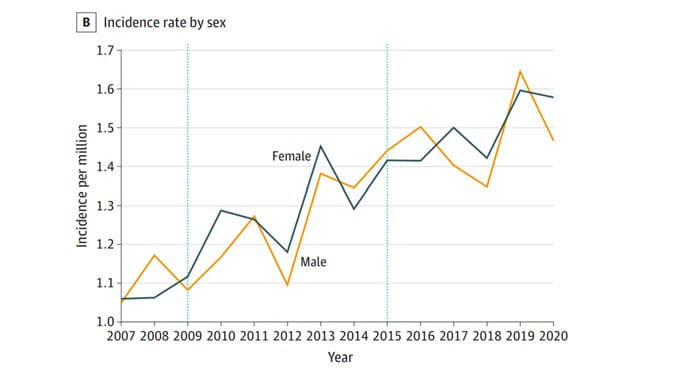
Note that we can’t tell whether these are sporadic CJD cases or variant CJD cases or even familial CJD cases; however, unless there has been a dramatic change in epidemiology, the vast majority of these will be sporadic.
The question is, why are there more cases?
Whenever this type of question comes up with any disease, there are basically three possibilities:
First, there may be an increase in the susceptible, or at-risk, population. In this case, we know that older people are at higher risk of developing sporadic CJD, and over time, the population has aged. To be fair, the authors adjusted for this and still saw an increase, though it was attenuated.
Second, we might be better at diagnosing the condition. A lot has happened since the mid-1990s, when the diagnosis was based more or less on symptoms. The advent of more sophisticated MRI protocols as well as a new diagnostic test called “real-time quaking-induced conversion testing” may mean we are just better at detecting people with this disease.
Third (and most concerning), a new exposure has occurred. What that exposure might be, where it might come from, is anyone’s guess. It’s hard to do broad-scale epidemiology on very rare diseases.
But given these findings, it seems that a bit more surveillance for this rare but devastating condition is well merited.
F. Perry Wilson, MD, MSCE, is an associate professor of medicine and public health and director of Yale’s Clinical and Translational Research Accelerator. His science communication work can be found in the Huffington Post, on NPR, and here on Medscape. He tweets @fperrywilson and his new book, How Medicine Works and When It Doesn’t, is available now.
Leave a Reply