
JULY 12, 2024
by Xia & He Publishing Inc.
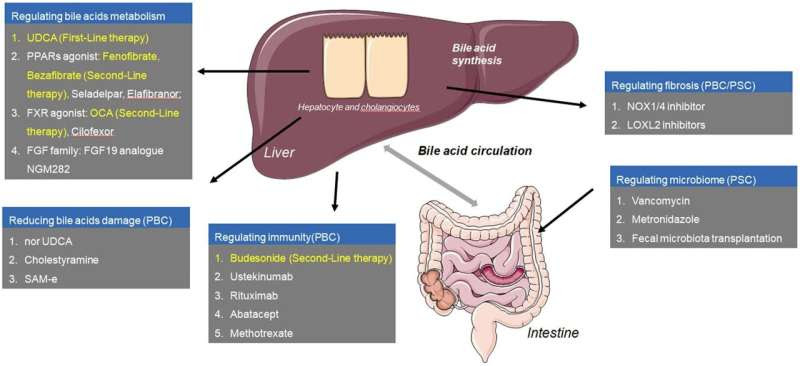
Graphical abstract. Credit: Journal of Clinical and Translational Hepatology (2024). DOI: 10.14218/JCTH.2023.00519
Cholestatic liver diseases (CLDs) are a group of disorders characterized by the impairment of bile formation, secretion, or excretion. This impairment leads to bile acid accumulation in the liver, causing liver biochemical abnormalities and histological damage.
Cholestasis can be either intrahepatic, involving damage within the liver bile ducts, or extrahepatic, involving obstruction of the bile ducts outside the liver. Chronic cholestasis can lead to liver fibrosis and cirrhosis. The most common types of CLDs are primary biliary cholangitis (PBC) and primary sclerosing cholangitis (PSC).
CLDs are classified into intrahepatic and extrahepatic types based on their etiology. Intrahepatic cholestasis is mainly caused by immune dysfunction, viral infections, drug-induced injury, and alcohol-related damage, whereas extrahepatic cholestasis often results from bile duct stones, tumors, or strictures.
Diagnosis typically involves evaluating elevated liver enzymes, specifically alkaline phosphatase (ALP) and gamma-glutamyl transferase (GGT). Elevated levels of both enzymes suggest cholestasis, with further imaging and endoscopic evaluations aiding in the diagnosis.
Additional diagnostic tools include liver biopsy, magnetic resonance cholangiopancreatography (MRCP), and endoscopic retrograde cholangiopancreatography (ERCP). These methods help in assessing the extent of liver damage and identifying the underlying cause of cholestasis.
The primary treatment of CLD focuses on alleviating the underlying cause and managing symptoms. Surgical or endoscopic intervention is effective for obstructive causes, while cessation of causative drugs or alcohol and antiviral treatments are recommended for drug-induced or viral-related cholestasis. The management of symptoms such as pruritus and fatigue is also crucial in improving the quality of life for patients with CLD. Symptomatic treatments include cholestyramine, rifampicin, and naltrexone for pruritus.
Pharmacological treatment of PBC include:
Ursodeoxycholic acid (UDCA): UDCA is the first-line treatment for PBC. It improves biochemical responses, delays histological progression, and extends transplant-free survival. The recommended dose is 13–15 mg/kg/day, with higher doses not showing additional benefits. UDCA works by inhibiting hydrophobic bile acid synthesis, enhancing bicarbonate secretion, and reducing cholangiocyte apoptosis. Despite its effectiveness, about 30–40% of PBC patients show an inadequate response to UDCA.
Obeticholic acid (OCA): For patients unresponsive to UDCA, OCA, a farnesoid X receptor (FXR) agonist, has shown efficacy in improving biochemical responses and delaying disease progression. However, it is associated with higher rates of adverse events like pruritus. OCA works by reducing bile acid synthesis and increasing bile acid transport from the liver.
Fibrates: Bezafibrate and fenofibrate, as adjunctive therapies to UDCA, have demonstrated improved biochemical responses in PBC patients with incomplete UDCA response. They work by activating peroxisome proliferator-activated receptors (PPARs), which inhibit bile acid synthesis. Clinical trials have shown that fibrates can significantly reduce ALP levels and improve patient outcomes.
Budesonide: In combination with UDCA, budesonide has shown potential in normalizing ALP levels in PBC patients. However, more extensive clinical trials are needed to confirm its benefits. Budesonide is a corticosteroid with high first-pass metabolism, which minimizes systemic side effects while targeting liver inflammation.
Currently, there are no approved pharmacological treatments for PSC. UDCA is used off-label, but its benefits are uncertain. Research is ongoing for potential new treatments including antifibrotic agents, immune-modulating therapies, and other novel drugs. Liver transplantation remains the only definitive treatment for advanced PSC, but recurrence of the disease post-transplant is common.
New treatment avenues for CLDs are being explored, including:
Fibroblast growth factor 19 (FGF19): Reduces bile acid synthesis and shows promise in early clinical trials for both PBC and PSC.
S-adenosyl-L-methionine (SAM-e): Enhances hepatocyte protection and may improve liver function in cholestatic conditions.
Steroid drugs: Anti-inflammatory properties help manage symptoms, particularly in autoimmune-related cholestasis.
Farnesoid X receptor agonists: Inhibit bile acid synthesis and promote bile flow, offering a new therapeutic target for various cholestatic conditions.
Significant progress has been made in understanding and managing cholestatic liver diseases. While UDCA remains the cornerstone of PBC treatment, new therapies like OCA, fibrates, and budesonide offer additional options for patients with poor responses to standard treatments. For PSC, research is ongoing to find effective treatments. Continued advancements in pharmacological therapies and better diagnostic criteria are crucial for improving patient outcomes in cholestatic liver diseases.
The management of cholestatic liver diseases requires a multidisciplinary approach involving hepatologists, gastroenterologists, and primary care physicians. Regular monitoring and early intervention can prevent complications and improve the prognosis for patients with these challenging conditions.
The findings are published in the Journal of Clinical and Translational Hepatology.
More information: Xin Luo et al, Progress in the Management of Patients with Cholestatic Liver Disease: Where Are We and Where Are We Going?, Journal of Clinical and Translational Hepatology (2024). DOI: 10.14218/JCTH.2023.00519
Provided by Xia & He Publishing Inc.
Leave a Reply